What are cilia?
Cilia are evolutionary well conserved, hair like structures on the surface of almost all mammalian cells. They are broadly divided into two categories: motile and non-motile cilia, the latter are also known as primary cilia.
Motile cilia occur in bundles of hundreds and are restricted to certain tissues such as respiratory epithelium or the reproductive system. They have a rhythmic waving pattern and are mainly involved in directional fluid movement and mucociliary clearance.
Primary cilia are solitary, non-motile antenna-like cell organelles on the surface of nearly all mammalian cells. They can transfer signals such as light, odor, directed flow or osmolarity gradients from nearby environment into the cell.
Even though mammalian cilia have already been described in the nineteenth century, their function remained unclear for many decades. In 1979 Afzelius described the correlation of situs abnormalities, chronic lung disease and the dysfunction of motile cilia (Primary Ciliary Dyskinesia, Kartagener Syndrome). In the 1990s the relationship between cystic kidney disease and ciliary dysfunction was discovered. Since then the scientific interest in primary cilia has risen dramatically. To date it is known, that primary cilia play a crucial role in many different biogical processes such as cell signaling, cell cycle, DNA damage repair pathways and embryogenesis. Many of the underlying intracellular mechanisms are still to be elucidated and of high scientific interest. A large number of inherited diseases have been associated with dysfunctional cilia. This group of diseases is summarized as “ciliopathies”.
Ciliary structure
The cilium consists of nine circular arranged microtubular doublets surrounded by a plasma membrane. While primary cilia are described to have a so called 9+0 structure, motile cilia contain an additional central doublet (9+2 Structure) as well as inner and outer dynein arms for ciliary motility.
Depending on the ciliary structure and function, three different types of cilia are described:
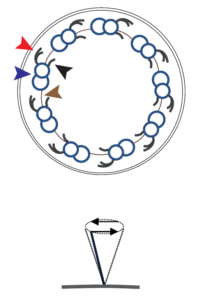
| Motile cilia (9 x 2 + 2 structure) occur in bundles of hundreds and produce a directional flow through rhythmic movements. Inner and outer dynein arms allow a directional ciliary movement by sliding of microtubular doublets against each other. Motile cilia are found in specialized tissues:
On respiratory cells for example they are responsible for a proper mucociliary clearance. In the oviduct they promote the transport of the zygote towards the uterus. Ependymal cells lining the brain ventricle produce a directional flow of the cerebrospinal fluid by ciliary movement. Also, the flagellum of mammalian sperm has a very similar structure (9×2+1) and ensures the motility of sperm. Dysfunction of these cilia cause, recurrent infection of respiratory tract and chronic lung disease, infertility as well as situs abnormalities.
|
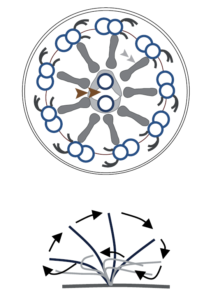 |
| Another motile, but solitary cilium is the primary cilium of the embryonal node. It is crucial for early embryonal development. Its 9 x 2 + 0 structure of nine microtubular doublets lack the central doublet but dynein arms ensure a microtubular movement. Altogether a rotatory beating pattern results out of this structure which is determining for a correct organ lateralization.
Dysfunction of this cilium causes heterotaxy or situs inversus.
|
 |
| Primary, non-motile cilia have a 9 x 2 + 0 structure. They lack the central pair as well as dynein arms. They can be found on nearly all mammalian cells and function as antennae for different signals into the cell. They are crucial for photoreception, olfaction, hearing and many intracellular pathways.
Dysfunction of this type of cilia causes diverse symptoms and syndromic ciliopathies associated with visual or hearing impairment, anosmia, renal, hepatic, cardiac and skeletal abnormalities. |
The cilium is anchored to the basal body from which it extends. Along the axoneme it can be structurally divided into following compartments: ciliary tip, ciliary body, ciliary necklace, transition zone, inversin compartment and basal body.
Cilia are surrounded by a ciliary membrane, the so called “transition zone” which acts as a barrier between the cilium and the rest of the cell where only a selective transfer of proteins can occur. Many gene mutations causing nephronophthisis result in a dysfunction of proteins in this “transition zone”.
Ciliogenesis
Ciliogenesis is tightly linked to the cell cycle (only in G1 and quiescence). After cell division the cilium is built from a so called “mother centriole”. Cilia cannot produce proteins themselves, thus all necessary components for ciliogenesis have to be carried into the cilium. The precise mechanisms are not yet fully understood. Some proteins are being transported via vesicles to the ciliary membrane and released there. The transport of proteins along the ciliary axoneme is a highly complex and energy dependent process, called “intraflagellar transport” (IFT). Defects in this process have been found to be causative for many ciliopathies such as Bardet Biedl Syndrome (BBS) or the group of short rib polydactyly syndromes.
Molecular ciliary functions
Cilia are involved in various essential molecular processes of the cell biology:
Cellcycle: Cilia evolve only in certain stages of the cell cycle. Also cells can only enter cell cycle when cilia are disassembled. Because of this dependence of processes it is considered, that cilia themselves have an influence on cell cycle regulation. This thesis is strengthened by different scientific results, even though the precise relationship between ciliary function and cell cycle remains to be elucidated.
Intraflagellar transport: Ciliary proteins have to be transported from the cell into the cilium an back via a complex, energy dependent process called “intraflagellar transport” (IFT), executed by different clusters of proteins. While “complex B” coordinates transporting proteins from the ciliary base to the tip (anterograde IFT), “complex A” is responsible for the retrograde IFT from the ciliary tip back to the base.
When IFT is inhibited in animal models, animals die in utero, which demonstrates the crucial role of this transporting process.
In humans, mutations in IFT- proteins cause defective development of the cartilage and bones. Those clinical patterns are summarized as “Short-Rib-Polydactyly Syndromes” (SRPS) including Jeune Asphyxiating Thoracic Dystrophy Syndrome (JATD), Mainzer Saldino Syndrome and different other. A connection between dysfunctional IFT and Bardet Biedl Syndrome has recently been found.
Ciliary signalling pathways
At least five different intracellular signalling pathways seem to play a significant role in the pathogenesis of different ciliopathies.
- Wnt-signalling: The Wnt-signalling pathway can be roughly divided into two branches: the “canonical” pathway which activates further target genes and the “non-canonical” pathway that plays a role in cell orientation. The influence of different ciliary proteins (e.g. NPHP3, NPHP4 etc.) on the Wnt-pathway was shown in animal models. The loss of these proteins results in developmental defects and cystic renal disease, presumably by the loss of adequate cell orientation.
- Hedgehog-signalling: This pathway is crucial for cartilage and bone development during embryogenesis and is IFT-dependent. Defective Hedgehog-signalling in mammals typically results in chondrodysplasia and bone deformities as well as complex developmental defects including polydactyly, heart defects, cystic renal disease or laterality abnormalities. The phenotypical spectrum comprises complex syndromes like “Jeune Asphyxiating Thoracic dysplasia”, “Mainzer Saldino Syndrome” or “Ellis van Creveld Syndrome”. Mutations in different ciliary genes (e.g. NPHP6, NPHP7, IFT140…) have been associated with a defective Hedgehog signalling pathway.
- Mamalian Target of Rapamycin (mTOR-)signalling: This pathway was shown to be activated in polycystic kidney disease (PKD). Inhibition of this pathway by the mTOR inhibitors Everolimus and Sirolimus lead to a significant reduction of cystogenesis and renal enlargement in animal models. Nevertheless, the effect of mTOR inhibition in human pharmacological studies was rather disappointing. Thus it remains unclear whether mTOR inhibition is a potential pharmacological target for cystic kidney diseases.
- Hippo-Signalling: The Hippo-pathway was found to play an essential role in organogenesis, cell regeneration and tumor suppression. Particularly its role in nephrogenesis and the development of the eyes has been pointed out. In animal models inactivation of the hippo-signalling pathway leads to polycystic kidneys and urinary concentrating defects, resembling the clinical phenotype of nephronophthisis. Mutations of NPHP genes such as NPHP3, NPHP4 and NPHP9/NEK8 result in an altered Hippo signalling suggesting a direct interaction. Still, exact mechanisms remain obscure.
- DNA-Damage Response: Recently the influence of NPHP4, NPHP9/NEK8, NPHP14 and NPHP15 on different processes of DNA-damage repair could be demonstrated.
Furthermore, ciliary proteins appear to be involved or interact with other essential mechanisms of cell biology such as intracellular calcium transport or the TGF-ß function. However, the exact mechanisms still remain to be elucidated.
What is a cyst?
Renal cysts are fluid filled cavities in the renal tissue that can appear in different number and size. Their clinical impact on the renal function can vary significantly. While unilateral, single cysts are most often accidentally found in routine ultrasound and are clinically irrelevant, in polycystic kidney disease multiple cysts occur bilaterally and lead to a progressive decline of renal function in most cases. Depending on the underlying disease, different segments of the nephron are involved in the development of cystic lesions: While in autosomal dominant polycystic kidney disease (ADPKD) cysts are found throughout the entire nephron, in other polycystic kidney diseases (ARPKD, NPH, BBS) they are limited to the collective duct. Both, number and size of renal cysts can progress during the course of disease and thereby replace healthy renal tissue. However, the development of a progressive loss of renal function is not only dependent on the size and quantity of cystic lesions but depends on the underlying disease as well as the individual clinical course.
From cyst to cilia?
The precise process of cyst formation is still not entirely understood. However, within the last 30 years tremendous progress has been made in the genetic unraveling and the molecular understanding of mechanisms involved in cystogenesis. Since the discovery of PKD1 as the genetic cause of ADPKD in 1985, more than 100 genes have been identified in which mutations lead to polycystic kidney disease. Moreover, at the end of the last century, it could be shown that the gene products of almost all those cystic genes were located at the so called cilia-centrosome complex, an antenna-like organelle protruding from almost all polarized cell types of the human body. Mutations of these genes resulted in an impaired ciliary function on the one hand and in the development of renal cysts on the other suggesting an essential pathophysiological role of cilia for cystogenesis. Consequently to date cystic kidney diseases are seen as the main representative of cilia-related-disorders called ciliopathies. But despite these major advances, the understanding of how defective cilia form renal cysts is still fragmentary. Thus further scientific efforts will be needed in order to obtain detailed insights into the molecular mechanisms of renal cyst formation and NEOCYST intends to play an essential part in this progress.
content:
Miriam Schmidts M.D. Ciliopathies: Their role in Pediatric Renal Disease