Erbliche zystische Nierenerkrankungen stellen als Gruppe eine der häufigsten genetischen Ursachen für Nierenversagen im Kindesalter dar. Die wichtigsten Vertreter dieser Krankheitsgruppe sind die Autosomal Rezessive Polyzystische Nierenerkrankung (ARPKD), Nephronophthise und Nephronophthise – assoziierte Ziliopathien (NPH/NPH-RC), Bardet – Biedl Syndrom (BBS) und HNF1B – Nephropathie (HNF1B). Es handelt sich jeweils um seltene Erkrankungen mit Inzidenzen von 1:5000 bis 1:100000. In Deutschland geht man davon aus, dass insgesamt ca. 300-450 Kinder betroffen sind. Die meisten dieser Erkrankungen werden autosomal – rezessiv vererbt. Eine Ausnahme stellt die HNF1B – Nephropathie dar, die autosomal dominant vererbt wird.
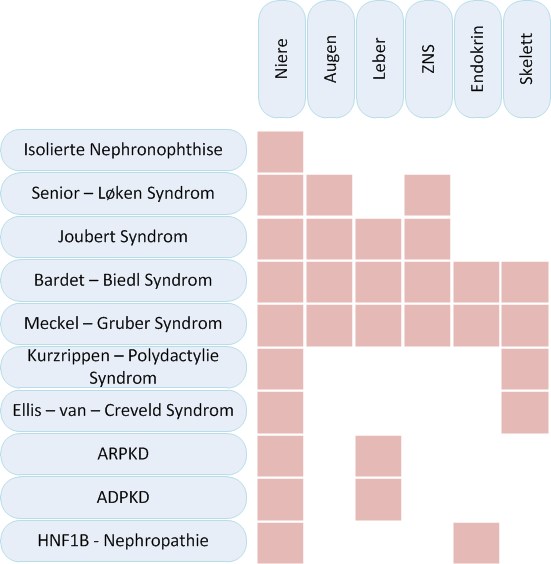
Zystische Nierenerkrankungen können isoliert die Niere betreffen oder mit Syndromen einhergehen die diverse weitere Organe betreffen können, insbesondere die Augen, Leber, endokrine Organe oder den muskuloskelettalen Apparat.
In den vergangenen Jahren konnten weit über 70 Gene identifiziert werden, deren Dysfunktion diese Erkrankungen verursachen können. Die meisten dieser Gene sind für die regelrechte Struktur und Funktion von primären Zilien verantwortlich. Primäre Zilien sind antennenähnliche Zellfortsätze, die vielfältige Aufgaben erfüllen. Sie sind essenziell bei Prozessen der Zellteilung, Zellorientierung, Fotorezeption, Mechanorezeption und Signalweiterleitung unter anderem in Wachstum und Entwicklung. (s. „Von Zilien und Zysten“)
Trotz des enormen wissenschaftlichen Fortschrittes sind die zugrundeliegenden Mechanismen der Krankheitsentstehung noch nicht vollständig geklärt.
Genetische und symptomatische Überlappungen
Es bestehen starke phänotypische und genotypische Überlappungen sowie eine ausgeprägte genetische Heterogenität der verschiedenen Krankheiten. So kann eine Erkrankung durch verschiedene Gene verursacht werden. Ebenso kann ein Gen verschiedene Krankheiten unterschiedlicher Ausprägung auslösen. Es können diverse Organe in unterschiedlichem Ausmaß betroffen sein. Der Beginn der Niereninsuffizienz kann stark zwischen Patienten mit dem gleichen betroffenen Gen variieren.
Die Diagnosestellung stellt daher eine große Herausforderung für behandelnde Ärzte und Betroffene dar.
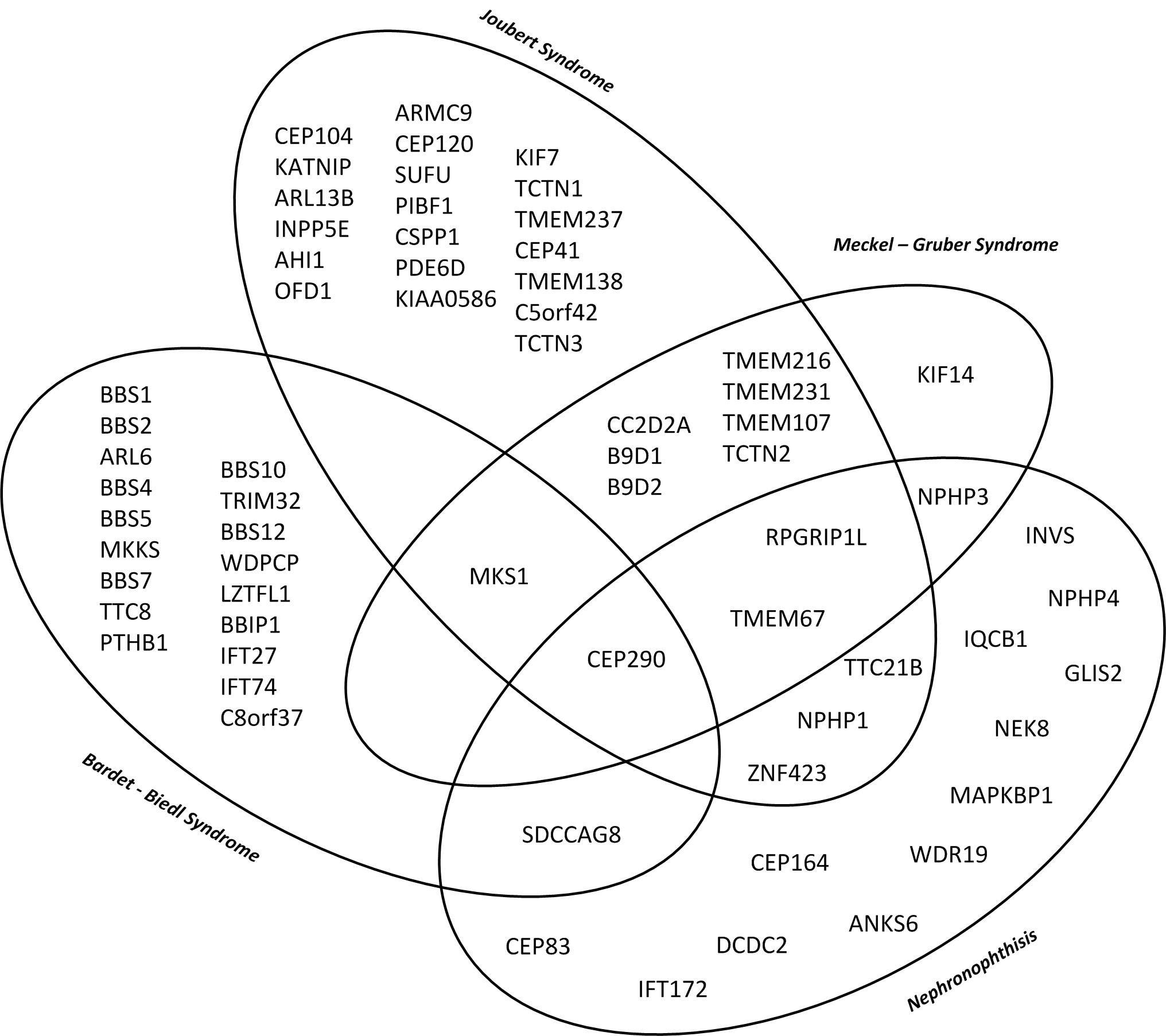
Dieses Diagramm zeigt die genetische und phänotypische Überlappung relevanter Ziliopathien mit Nierenbeteiligung von der isolierten Nephronophthise bis zum letalen Meckel – Gruber Syndrom mit multipler Organbeteiligung. (10)
 |
 |
Bisher bekannte Gene hereditärer zystischer Nierenerkrankungen
| Nephronophthise (1,2)
(~63% genetisch gesichert) |
>1 % der Fälle | < 1 % der Fälle |
|
|
|
| Joubert Syndrom (3)
(62 – 94% genetisch gesichert) |
>1 % der Fälle | < 1 % der Fälle |
|
|
|
| Bardet – Biedl Syndrom (4)
(70-80% genetisch gesichert) |
häufigste Gene | seltenere Gene |
|
|
|
| Meckel – Gruber Syndrom (5,6)
(60% genetisch gesichert) |
häufigste Gene | seltenere Gene |
|
|
|
| Senior – Løken Syndrom |
|
|
| Kurzrippen – Polydaktylie Syndrom – incl. Mainzer – Saldino Syndrom (MZSDS) und Jeune asphyxierende Thoraxdysplasie (ATD) (7) |
|
|
| Ellis van Creveld Syndrom |
|
|
| Autosomal Rezessive Polyzystische Nierenerkrankung – ARPKD (8) |
|
|
| Autosomal Dominante Polyzystische Nierenerkrankung – ADPKD (9) |
|
|
| HNF1B – Nephropathie |
|
|
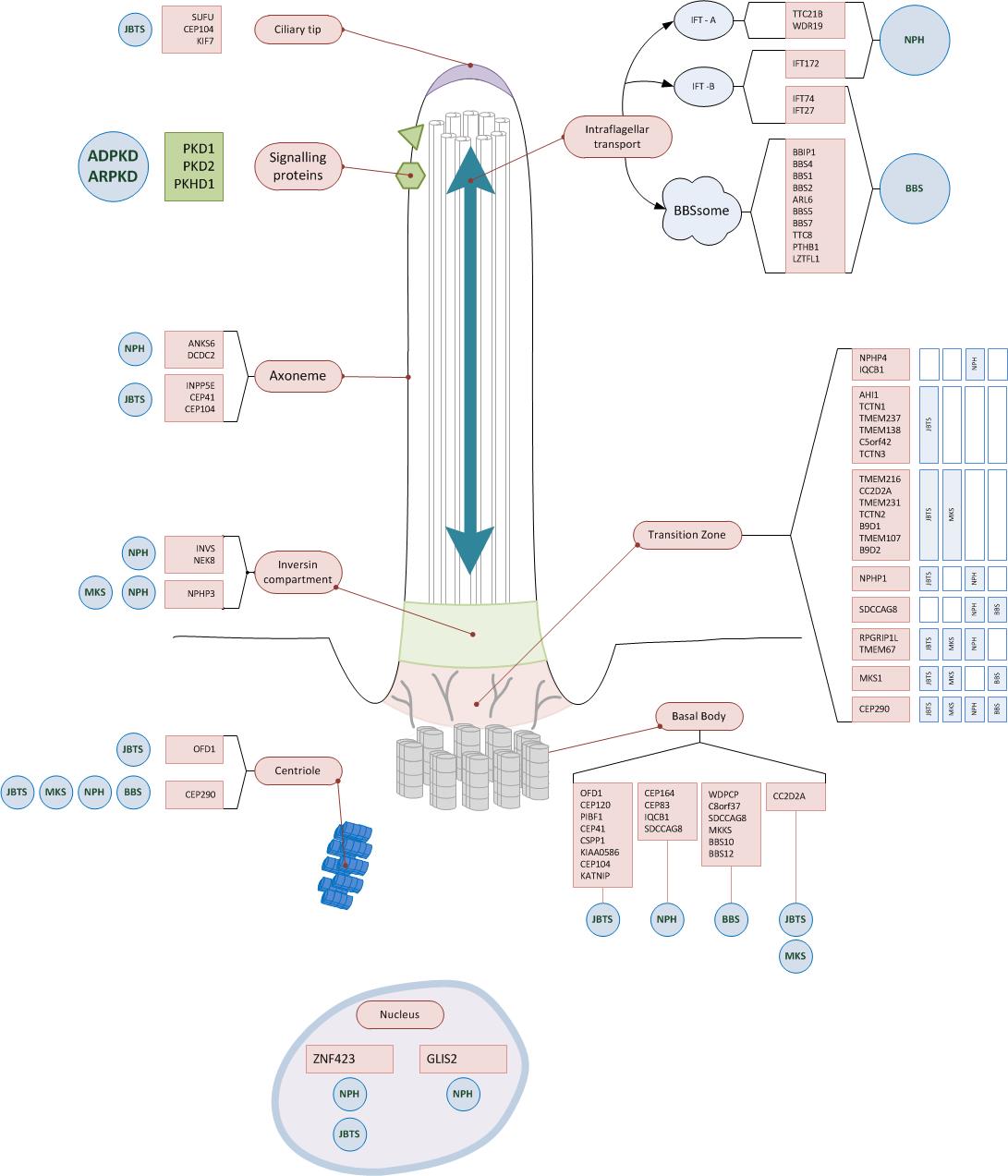
Betroffene Strukturen am primären Zilium
Die meisten Gene die bei zystischen Nierenerkrankungen betroffen sind, kodieren für Proteine am primären Zilium. Häufig erfüllen sie Funktionen im Bereich der Transitionszone, des Basalkörperchens oder beim intraflagellären Transport. (10)
Quellen (1) Stokman M. et. al GeneReviews® [Internet]. http://www.ncbi.nlm.nih.gov/books/NBK368475/ PMID: 27336129. (2) König J. et. al, Phenotypic Spectrum of Children with Nephronophthisis and Related Ciliopathies. Clin J Am Soc Nephrol. 2017 Dec 7;12(12):1974-1983. PMID: 29146700; (3) Parisi M. et. al, GeneReviews® [Internet]. http://www.ncbi.nlm.nih.gov/books/NBK1325/ PMID: 20301500. (4) Ece Solmaz A. et. al, Targeted multi-gene panel testing for the diagnosis of Bardet Biedl syndrome: Identification of nine novel mutations across BBS1, BBS2, BBS4, BBS7, BBS9, BBS10 genes. Eur J Med Genet. 2015 Dec;58(12):689-94. PMID: 26518167. (5) Hartill V. et. al, Meckel-Gruber Syndrome: An Update on Diagnosis, Clinical Management, and Research Advances. Front Pediatr. 2017 Nov 20;5:244. PMID: 29209597; (6) Szymanska K. et al, Founder mutations and genotype-phenotype correlations in Meckel-Gruber syndrome and associated ciliopathies. Cilia. 2012 Oct 1;1(1):18. PMID: 23351400; (7) Braun DA, Hildebrandt F. Ciliopathies. Cold Spring Harb Perspect Biol. 2017 Mar 1;9(3). pii: a028191. PMID: 27793968; (8) Lu H. et. al, Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat Genet. 2017 Jul;49(7):1025-1034. PMID: 28530676; (9) Cornec-Le Gall E. et. al, Genetic Complexity of Autosomal Dominant Polycystic Kidney and Liver Diseases. J Am Soc Nephrol. 2018 Jan;29(1):13-23. PMID: 29038287; (10) König JC. et. al, Network for Early Onset Cystic Kidney Diseases – A Comprehensive Multidisciplinary Approach to Hereditary Cystic Kidney Diseases in Childhood. Front Pediatr. 2018 Feb 13;6:24. PMID: 29497606; (11) www.omim.org